
Insofar as proof to 100% certainty does not exist, what is meant by the term ‘proof’ is in fact a sufficiency of data to support arriving at a given conclusion. ‘Evidence’ is simply the information and data, from which we form a conclusion. Whether it is ’sufficient’ is dependent on what we already know, and what we are seeking to establish, relative to the models of empirical validation that exist [i.e., cohort study vs. RCT]. ‘Proof’ of causation is therefore entirely relative, and different levels of ‘sufficiency’ may satisfy different purposes, where the ultimate standard of proof is the ability to make connections and conclusions from the available evidence.
However, incorrect assumptions about causation together with methodological prejudice has generated a number of issues for nutrition science, and a dismissal of the processes through which we arrive at conclusions based on the evidence. Both of these issues will be discussed in this second part of this article.
3) Methodolatry results in incorrect assumptions about RCTs and causation
As discussed in Part 1, the limitation for nutrition from a ‘cause-effect’ perspective is that the RCT was designed to test pharmaceuticals, i.e. single compounds with a targeted pharmacokinetic and pharmacodynamic profile. This design does not always adequately translate the effect of a single nutrient on a single endpoint into an accurate reflection of the effect of a complex food matrix of multiple constituents, and an overall diet pattern, on multifactorial disease processes.
Perhaps more problematic for the wider nutritional discourse is the assumption that any results from a trial labelled an ‘RCT’ automatically reflect causation. This is simply incorrect. Even leaving aside assumptions and presuppositions underpinning strong internal validity, any trial demonstrated a cause-effect relationship simply shows that intervention A caused outcome B given the circumstances/settings/characteristics in which the intervention was conducted (1).
This is fundamental, because the net effect is that evidence can be overstated or understated depending on what purpose – what research question and hypothesis – an RCT sought to test and answer, and what methodological limitations it may have. Yet, there is a default presumption that the results of any RCT indicate a cause-effect relationship. In the context of nutrition this presumption is often untenable. As the null hypothesis of an RCT on a nutrient supplement is, “Nutrient A has no effect on health outcome B”, the inevitable effect of affirming the null is to imply that there is no effect of a nutrient, when we know as a matter of empirical fact that all nutrients have biological activity and influence health status (2).
In fact, the reductionist approach of trialling isolated nutrients in RCTs is an expensive example of what results when an RCT tests the wrong research question. The null findings of numerous large scale trials on antioxidants, vitamin E, or long-chain omega-3’s, do not reflect that the nutrient has no effect on health outcome X. The actual hypothesis being tested was whether an isolated supplemental – often synthetic – form of a nutrient has the same biological effect as the nutrient consumed in the context of a total diet pattern. Or, whether a certain intake level of nutrient X in the context of baseline level Y increases/decreases risk of health outcome Z. Instead, many of these trials failed to ask the right research question, testing only ’supplement dose X’ without regard to the obvious fact that nutrients only ever act within biological range, and thus their effects are always relative to the non-linear bell curve which defines the health effects of nutrients (3).
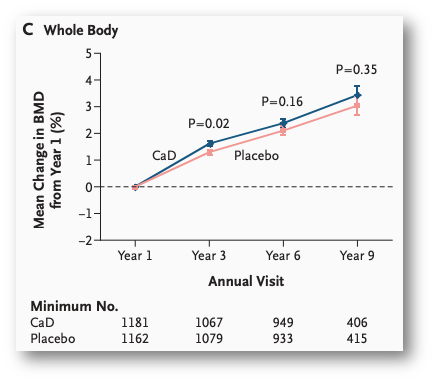
Figure from Jackson et al. (N Engl J Med. 2006;354:669-83.), a “gold standard”, randomised, double-blind, placebo-controlled intervention comparing 1,000mg calcium + 400IU vitamin D to placebo for effects on bone mineral density. ‘No significant difference’ after 9yrs, however, the “placebo group” had mean total calcium intake similar to the intervention group, and 65% had at least >800mg/d calcium intake. Null by design.
The reality is that any model of empirical validation is only as good as the question asked, the hypothesis tested, and the assumptions underpinning the design. The traditional strength of the RCT is that it may identify causal relationships, but this is a contingent capacity, not a biomedical birthright. This latter play on words is not random, because the biomedical model gave birth of the full explanatory RCT as we currently have it, with internal validity contingent upon having exchangeability of participants at the subgroup level, a clearly defined intervention and placebo, and identifiable independence of effects (4). As discussed in Part 1, many of these criteria are practically impossible to achieve for nutrition, with the net effect that any result from an explanatory RCT does not in point of fact demonstrate causation from a validity standpoint, even if the findings were positive.
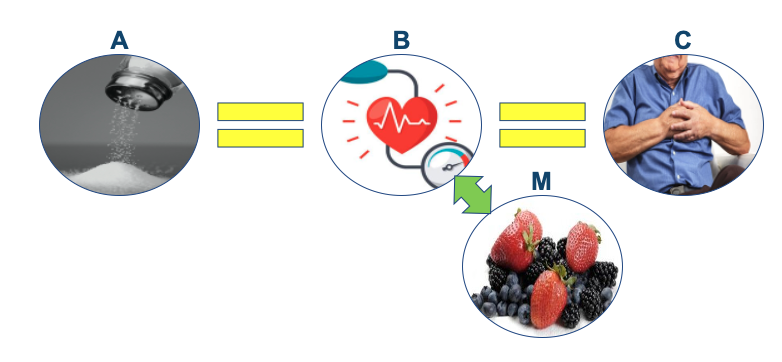
More importantly for nutrition science is the fact that when it comes to diet-disease relationships, there is simply no such thing as A = B. All diet-disease relationships are mediated by the effect of diet, total or specific characteristics thereof, on physiological processes. The relationship between added sugar and liver fat accumulation is mediated by impacts on, amongst others, hepatic insulin resistance and increased de novo lipogenesis. The relationship between saturated fat and heart disease is mediated by impacts on, amongst other variables, LDL-cholesterol. Thus, all diet disease relationships are A = C via B. We can even take this equation further because the influence of diet on ‘B’ is itself contingent on the fact that no characteristic of diet exists in isolation, and there is always a relationship between a given characteristic and other constituents of diet. These factors are referred to as ‘moderating’, ’supporting’, or ‘mediating’ factors [the terms are interchangeable], but may be crucial to the operation of any cause-effect relationship (5). The equation then becomes: A = C via B(M), where ‘M’ refers to the mediating factors that influence the operation of the causal relationship between A and C.
Salt, hypertension, and CVD provides another example of this relationship where high sodium diets (A) causally increase risk for heart disease and stroke (C), via increased hypertension and associated adverse physiological effects (B), a relationship which may be mediated by potassium intake, flavanoid intake, age, and other factors like stress (M). The crucial thing to note in this case is that any attenuation of the relationship between A and C through M does not invalidate that a causal relationship between A and C exists. This is an error in reasoning that is often made when looking at diet-disease relationships, because again, nothing in diet exists in a vacuum. It doesn’t invalidate the relationship because none of the mediating factors could in and of themselves have a causal relationship with C.
These factors are fundamental to the inferences that many are quick to make about the results of an RCT. The assumption that randomisation itself controls for all unknown variables, or even known variables, in the case of nutrition; for example, the influence of baseline nutrient status on the outcome of interest, which is often never even stratified or taken into account. Or the assumption that the effect size is the ‘true’ effect size, particularly where that is then compared to the effect size observed in epidemiology, and the presumption in favour of the result from the former that it is more reliable, and ‘true’. The assumption that adjusting for mediating factors leaves us with more accurate results; rather than consider effect modifiers, all moderating factors are treated as confounders, which can often result in over-adjustment in statistical models. The numerous studies on saturated fat and heart disease that have adjusted for blood cholesterol and LDL-cholesterol levels serves as a case in point for over-adjustment.
The reality is all of these assumptions are shaky when we factor in the indirect causal nature of diet-disease relationships. It cannot be taken that simply because a trial uses an RCT design that the results from that population subgroup are:
a) automatically correct;
b) applicable in that population in the real-world (unless the trial was done in the real world, in which case it is ‘pragmatic’ and strong internal validity is less of a strict policy), or;
c) by default a more accurate estimate of effect than observational research.
In their extensive essay critique of RCTs, Deaton & Cartwright (1) stated the following:
“RCTs are oversold when their non-parametric and theory-free nature, which is arguably an advantage in estimation or internal validity, is used as an argument for their usefulness. The lack of structure is often a disadvantage when we try to use the results outside of the context in which the results were obtained; credibility in estimation can lead to incredibility in use. You cannot know how to use trial results without first understanding how the results from RCTs relate to the knowledge that you already possess about the world, and much of this knowledge is obtained by other methods. Once RCTs are located within this broader structure of knowledge and inference, and when they are designed to enhance it, they can be enormously useful, not just for warranting claims of effectiveness but for scientific progress more generally. Cumulative science is difficult; so too is reliable prediction about what will happen when we act.”
The TL;DR here is that there is too much reliance in evidence-based discourse on the design per se as indicating causation, when the ability of an RCT to do is a rebuttable presumption upon critique of the research question asked, the underlying assumptions and presuppositions for validity, and background knowledge underpinning the use of the design.
Good causal inference is a process, and may be deduced from different models of empirical validation, through critical appraisal of the overall existing evidence-base. Assuming the results of an RCT by default equal causation is not only intellectual indolence, it is incorrect.
4) Methodological prejudice takes precedent over the process of evaluation
The ultimate standard of proof in science is the ability to make connections and conclusions from the available evidence. The is a process of evaluation, which can be stifled by methodological prejudice, a term for which Deaton & Cartwright (1) warrant a blockquote in relation to this issue:
“Contrary to much practice in medicine as well as in economics, conflicts between RCTs and observational results need to be explained, for examples by reference to the different characteristics of the different populations studied in each, a process that will sometimes yield important evidence, including on the range of applicability of the RCT results themselves. While the validity of the RCT will sometimes provide an understanding of why the observational study found a different answer, there is no basis (or excuse) for the common practice of dismissing the observational study simply because it was not an RCT and therefore must be invalid. It is a basic tenet of scientific advance that, as collective knowledge advances, new findings must be able to explain and be integrated with previous results, even results that are now thought to be invalid; methodological prejudice is not an explanation.”
A classic example of methodological prejudice, where study design outranked reconciling the evidence by further analysis, can be seen in the observational research finding an association between post-menopausal women taking hormone replacement therapy had reduced risk of cardiovascular disease (6). A subsequent RCT on the issue found that women taking HRT had increased risk of CVD. Given the status of the RCT over a cohort study in the evidential pyramid, the immediate assumption was that the observational study had got it wrong, and the RCT was correct. In fact, further analysis revealed that both studies had been in different patient populations, and both were correct for their respective population subgroups whom had been studied. The discrepancy related the discrepancy between the timing of the onset of menopause and initiation of HRT. Where these factors were accounted for, both the observational cohort study and the RCT yielded similar results.
An example from nutrition sciences can be observed in the discrepancy in the associations between dietary vitamin E intake and neurodegenerative disease in epidemiology, and overall lack of effect of vitamin E in RCTs. The observations in nutritional epidemiology are consistent, observed in different populations, and supported by mechanistic studies, including brain autopsy studies of brain vitamin E content in deceased Alzheimer’s disease persons (7,8). However, numerous RCTs of supplemental vitamin E and cognitive decline have failed to find an effect (9,10). The RDA for vitamin E is 15mg/d, and post-hoc analyses of interventions revealed that an effect of supplementation was only observed in participants with baseline levels of vitamin E <40% of the RDA [<6.1mg/d] (9,11). Had trials only included subjects with low baseline levels, uniformity may have been more achievable – and results more consistently positive.
A further issue that complicates this presupposition is that consumption of a supplemental nutrient later in life – where the pathophysiology of a condition is more advanced – is not evidence of the benefit of consuming that nutrient through sustained dietary intake earlier in life. In addition, many supplement trials use synthetic alpha-tocopherol, one of eight isoforms of tocopherols and tocotrienols that form vitamin E in nature; of particular relevance is that brain autopsy studies have revealed gamma-tocopherol is higher in brains of persons who died with a healthy brain (8). Methodological prejudice dictates a conclusion of ‘vitamin E has no effect on cognitive decline’, a conclusion which is as inconsistent with biological plausibility as it is with wider support from other lines of inquiry. The reality is that discrepancy in relation to this research question arises primarily from RCTs; arguable on balance of evidence, it is the RCTs yielding incorrect answers. Answering this question correctly, to the closest approximate truth that is possible, comes back to the overall process of evaluation, rather than defaulting to an assumption that the results of the RCTs reflects that ‘truth’.
The issue may be considered one of inflexibility, an over-reliance on the hierarchy of evidence as a standard of proof, when both frameworks are separate and distinct. The process of establishing causal inference is a process of overall evaluation, where the standard of ‘proof’ to be met is always relative. In this respect, relying on a ranking system that only reflects increasing confidence in results from a causation perspective is a self-limiting exercise in critical appraisal, when [for reasons outlined above and in Part 1] causation may not even be demonstrable in trials nominally carrying the label of an RCT. It misplaces the use and value of respective study designs, while potentially understating or overstating the results from a given study merely due to its design. These are the potential limitations of methodological prejudice, the evidential hierarchy interpreted inflexibly as an inanimate and static structure for what is a dynamic, evolving, multilayered process of scientific evaluation. In this dynamic process, each design has merit and differentially contributes to informing the overall evaluation of evidence. Consider that discordant findings in epidemiological research often lead to a magnification of criticism of observational research; discordant findings in RCTs, on the other hand, never result in the ‘gold standard’ designation even being questioned. Inconsistencies are “explained away or swept under the rug” (12).
It is commonly overlooked that public health policy is often formed and applied through multiple lines of evidence not including RCTs. It is a process of evaluation, where the hierarchy of evidence informs the evaluation of the research question but is not considered sacrosanct, and where there is no presumption that a research question can only be answered by an RCT. For public health practice [which includes private practice], it is a process in which we aim to base our decisions on the highest quality evidence available. However, this statement on its own tends to lead to methodological prejudice based on the the association between “quality” and “highest on the evidential pyramid”. In reality what highest quality evidence available means is the best information we have at the present time that supports an action to achieve a beneficial outcome. The ability to make conclusions based on the available evidence is seldom supported by one line of evidence alone. Myopic focus on one over others overlooks the fact that any given design is merely a lens through which to view the overall evidential picture, but this does not always mean the lens is focused correctly or on the right issues.
This analogy of a focused lens is important for considering scientific inquiry into modern diet-disease relationships, which unlike the early nutrient-deficiency focus of nutrition science, do not have single-compound pathways, short latency periods and rapid recovery when addressed; they are characterised by long latency periods, often over 3 to 4 decades, and complex aetiology. Focusing the lens in on RCTs alone can lead to a failure to see the forest for the trees, and seeing the forest is nearly always a case of incorporating converging lines of evidence. Echoing Deaton and Cartwright, there is simply no basis for dismissing a study or multiple lines of inquiry on the basis that there is no RCT, or a given study was not an RCT.
Conclusion
The first point is a word of caution that this essay [through both parts] should not be taken as a broad dismissal of RCTs. They have their place in the overall process of scientific evaluation. Rather, the purpose of this essay was to highlight some of the fundamental differences that those of you interpreting nutrition research should bear in mind, and to caution against the oversimplification of the process of critical evaluation that results from placing unwarranted emphasis, and often blind faith, in the ‘correctness’ of RCTs. This is a caution against naive omission of the processes of interpretation, synthesis of multiple lines of evidence, and appropriate extrapolation, that occurs from overly simplistic differential values attached to different sources (13).
As an early proponent of the evolution of ‘evidence-based medicine’, Austin Bradford-Hill once stated that “any belief that the controlled trial is the only way to conduct research would mean not that the pendulum had swung too far but that it had come right off the hook” (14). Given the Bradford-Hill criteria for evaluating causation remains as cogent today as it was in 1965, students of health sciences would be well served to sharpen their critical thinking skills on such a tool than be seduced by the simplicity of relying on the hierarchy of evidence. To quote again from Deaton & Cartwright (1):
“The gold standard or ‘truth’ view does harm when it undermines the obligation of science to reconcile RCTs results with other evidence in a process of cumulative understanding.”
There is more to scientific thinking then ‘was the trial randomised?’.
_________________________________________________________________________________
References:
1. Deaton A, Cartwright N. Understanding and misunderstanding randomized controlled trials. Soc Sci Med. 2018;August(210):2-21.
2. Blumberg J, Heaney R, Huncharek M, Scholl T, Stampfer M, Vieth R et al. Evidence-based criteria in the nutritional context. Nutrition Reviews. 2010;68(8):478-484.
3. Heaney R. Nutrients, Endpoints, and the Problem of Proof. The Journal of Nutrition. 2008;138(9):1591-1595.
4. Zeilstra D, Younes J, Brummer R, Kleerebezem M. Perspective: Fundamental Limitations of the Randomized Controlled Trial Method in Nutritional Research: The Example of Probiotics. Advances in Nutrition. 2018;9(5):561-571.
5. Suzuki E, VanderWeele T. Mechanisms and uncertainty in randomized controlled trials: A commentary on Deaton and Cartwright. Social Science & Medicine. 2018;210:83-85.
6. Vandenbroucke J. The HRT controversy: observational studies and RCTs fall in line. The Lancet. 2009;373(9671):1233-1235.
7. Morris M, Evans D, Tangney C, Bienias J, Wilson R, Aggarwal N et al. Relation of the tocopherol forms to incident Alzheimer disease and to cognitive change. The American Journal of Clinical Nutrition. 2005;81(2):508-514.
8. Morris M, Schneider J, Li H, Tangney C, Nag S, Bennett D et al. Brain tocopherols related to Alzheimer’s disease neuropathology in humans. Alzheimer’s & Dementia. 2015;11(1):32-39.
9. Kang J, Cook N, Manson J, Buring J, Grodstein F. A Randomized Trial of Vitamin E Supplementation and Cognitive Function in Women. Archives of Internal Medicine. 2006;166(22):2462.
10. Kang J, Cook N, Manson J, Buring J, Albert C, Grodstein F. Vitamin E, Vitamin C, Beta Carotene, and Cognitive Function Among Women With or at Risk of Cardiovascular Disease. Circulation. 2009;119(21):2772-2780.
11. Morris M, Tangney C. A Potential Design Flaw of Randomized Trials of Vitamin Supplements. JAMA. 2011;305(13):1348.
12. Concato J, Horwitz R. Randomized trials and evidence in medicine: A commentary on Deaton and Cartwright. Social Science & Medicine. 2018;210:32-36.
13. Swales J. Evidence-based medicine and hypertension. Journal of Hypertension. 1999;17(11):1511-1516.
14. Hill A. Heberden Oration, 1965. Reflections on the Controlled Trial. Annals of the Rheumatic Diseases. 1966;25(2):107-113.